Maebashi Institute of Technology Department of Life Science and Informatics
Computational Life Science and Chemistry Research Group
We are conducting analysis of biological properties by developing computer programs. Due to the rapid development of molecular biology in recent years, information regarding life science, such as DNA sequence and structure of biopolymers is rapidly increasing. However, we are still far from fully exploiting this information for practical applications. We are aiming to extract new knowledge from these data by using computers. Followings are a couple of examples of our study.
Profile Analysis of Next Generation Sequencer
Most biological functions in our body depend on proteins, which is a linear polymer of 20 different kinds of amino acids.
The sequential order of amino acids is coded as nucleotide sequence in the genome DNA.
By reading the base sequence of whole genome DNA sequence,
we can learn the information of all functioning proteins, in a way.
Although it takes another large step to really understand what the sequence means,
it is at least an important first step to understand the life system.
In 2001, the first human draft genome sequence (3 billion base pairs, about 700 MB of information)
was declared to be completed.
It required effort of international consortium for almost ten years, and cost about 3 billion dollars.
Genomes of hundreds of species have been sequenced so far.
However, we need to read more sequences because there are much more different species,
and also because each individual among human being has slightly different sequence that determines
the difference of each person.
A new technology called Next Generation Sequence (NGS) emerged during last five years.
With this method, genome DNA is broken down to small pieces (ca. 30-100 base pairs), and these short sequences are
put together using high performance computers.
They say with NGS, a full genome sequence of a person can be analyzed with less than 1,000 dollars.
One major drawback of NGS is, very often it is not so straightforward to assemble the short reads into one complete sequence.
There are three major platforms of NGS, Solid, Roche 454, and Illumina.
Among others, Illumina is dominating the market due to its high performance.
During the course of analyzing data from our collaborator,
we identified the sequence specific error profile of Illumina sequencer.
We also found this error profile is one of the major cause of the difficulty during assembling and identification of mutations.
Based on this knowledge, we are now trying to develop methods to circumvent the problem.

The mapping result of Illumina reads on genome sequence along the horizontal axis. The red dots indicate the sequencing error. They are obviously concentrated on some specific regions of the genome.
Molecular Evolution of Multicopper Oxidases
Small amount of minerals are essential for the life system. They say more than half of the all proteins function interacting with metal ions. We made molecular evolution analysis of proteins involving copper ions. The metal binding protein suits the sequence analysis because it requires specific amino acids at specific sequence positions in order to properly hold metal ions. In case of copper proteins, cystein and histidine are the amino acids to be conserved. As a result, we proposed an evolution model of known multicopper oxidases (Nitrite reductase, Laccase, and Ceruloplasmin) and predicted the presence of three evolutionary intermediates ([A], [B] and [C]) as described in the figure below. Also, we discovered many sequences corresponding to the evolutionary intermediates do exist in genome sequence database of contemporary organisms. We then proposed these evolutionary intermediates should be taking homo trimer structures based on the inference from our evolution model. Later on, four independent research groups reported the X-ray structures of these intermediate proteins as homo trimers, in accordance with our prediction.
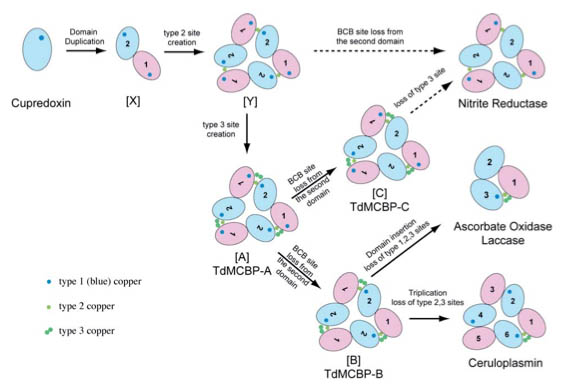
Predicted evolution model of multicopper oxidases. Three proteins on the right column (Nitrite reductase, Laccase, and Ceruloplasmin) have been known. The presence of three evolutionary intermediate proteins ([A], [B] and [C]) was proposed. All proteins described here are expected to be evolved from the monodomain cupredoxin at the top left corner.